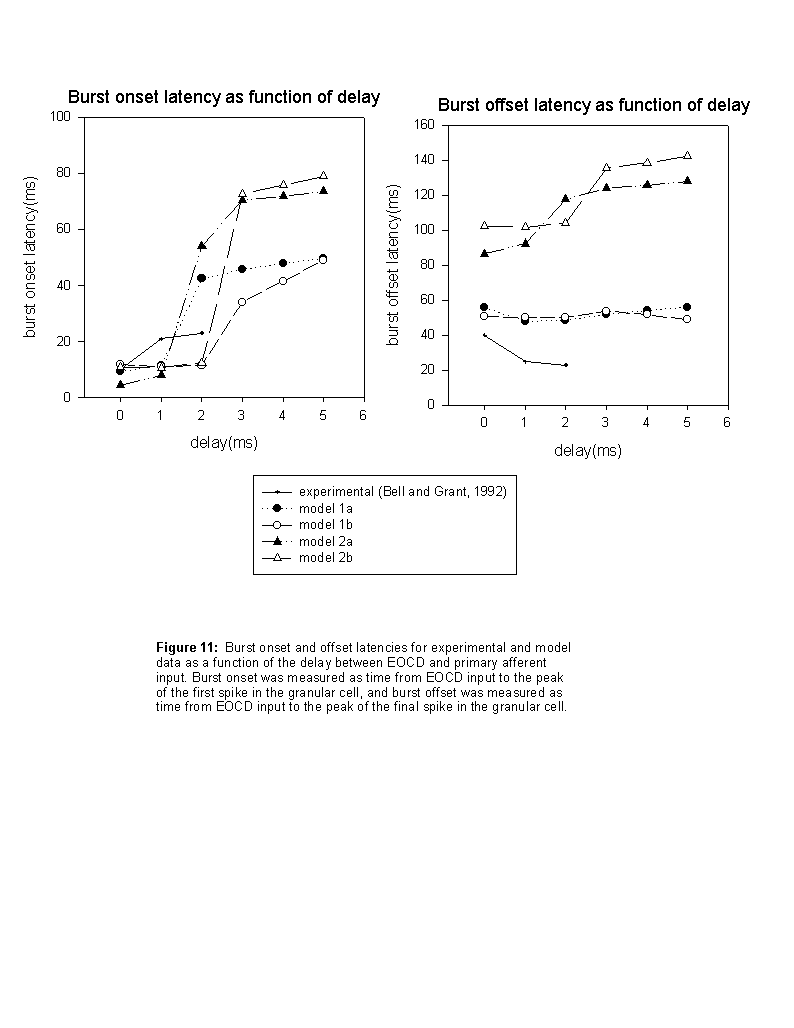
Intrinsic control of burst offset
Although the model was successful in describing the increase in the latency from the juxtalobar input to the first granular cell spike as the delay between the two inputs increased (figure III-11-i), the corresponding decrease in burst offset latency shown by extracellular field recordings from the granular cell layer (figure III-2b) was fit poorly by both models of type 1, with LMI controlled burst offsets, and type 2, with intrinsically ending bursts (figure III-11-ii). However, each of these models suggests possible solutions to this discrepancy.
Models 2a and 2b showed that the gradual rise of the slow, noninactivating potassium current, I-KM, was necessary for intrinsic cessation of bursting. Examination of contour plots describing spike number output of the model with varying levels of the maximum conductances for both I-KM and I-NaP revealed that reducing I-KM to zero resulted in uncontrolled repetitive firing over a broad range of values for I-NaP. This predicted transition to repetitive firing could be tested in vitro under appropriate input conditions by application of Cs+ and/or Ba2+ to chemically block I-KM channels, along with application of picrotoxin to block GABAA mediated inputs originating from ephaptic stimulation of LMI presynaptic terminals.
It is possible, however, that the gating kinetics of the I-KM mediated current in ELL granular cells differ from those reported in D’Angelo et al. (2001) for rat cerebellar granule cells. Specifically, this model suggests that an increase in the activation kinetics of I-KM over a similar voltage range would accelerate burst offset by overtaking the depolarizing effects of I-NaP mediated inward current. Furthermore, activation of I-KM at slightly more depolarized membrane potentials would allow for burst onset latency to more dramatically influence burst offset latency; longer periods of subthreshold depolarization would then significantly increase the proportion of I-KM channels that were already open at the onset of the burst, thus decreasing the number of spikes that could be produced before I-KM could successfully antagonize the regenerative effects of both I-NaP and I-NaR. These properties of the gating kinetics for I-KM could be determined by patch clamping granular cells in vitro in the presence of tetraethylammonium (TEA) and chemical agents that selectively block other K+ channel subtypes while leaving I-KM mediated current intact. Modeling of patch clamp results would then allow characterization of the exponential functions describing kinetics for I-KM.
Another possible mechanism for intrinsic control of burst offset suggested by the model involves the I-KCa / I-CaHVA system. The effect of this system in the present model was limited to minor spike amplitude adaptation and spike number response tuning. However, it is conceivable that intracellular calcium accumulation during the burst, especially in areas containing dense clusters of I-KCa channels, could allow large, hyperpolarizing effluxes of K+ to overcome regenerative Na+ currents and prevent subsequent spikes. Control of this effect by input delay would need to be achieved via Ca2+ influx at subthreshold membrane potentials, exerting a burst onset latency-dependent effect similar to that described above for the putative role of I-KM.
Although low-voltage activated Ca2+ channels have been described in immature rat cerebellar granule cells in situ (D’Angelo, Filippi, Rossi, and Taglietti, 1997), the subthreshold Ca2+ current carried by these channels, mainly involved in Ca2+ spikes at an early stage of development, was almost nonexistent in mature cells. In any case, Ca2+ channels in ELL granular cells are remain uncharacterized. In vitro patch clamp examination of calcium conductance at subthreshold membrane potentials in ELL granular cells, in concert with future modeling of more complete calcium dynamics (e.g. release from intracellular compartments, possible localization at I-KCa channel clusters, or buffering), would allow testing of this hypothesis.
{kind=link}
{kind=link}